
Deciphering the m6A code via quantitative profiling of m6A at single-nucleotide resolution
Published in bioRxiv, 2019
Recommended citation: Garcia-Campos, M.A., Edelheit, S., Toth, U., Shachar, R., Nir, R., Lasman, L., Brandis, A., Hanna, J.H., Rossmanith, W. and Schwartz, S., 2019. Deciphering the "m6A code" via quantitative profiling of m6A at single-nucleotide resolution. BioRxiv, p.571679. https://www.biorxiv.org/content/10.1101/571679v1
Abstract
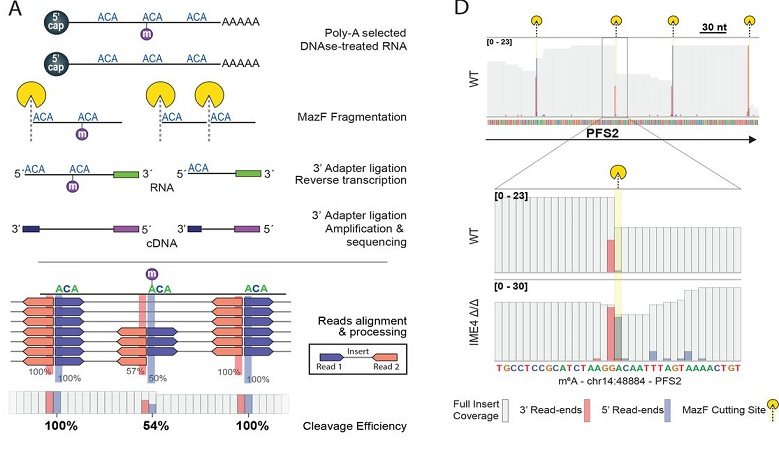
N6-methyladenosine (m6A) is the most abundant modification on mRNA, and is implicated in critical roles in development, physiology and disease. The ability to map m6A using immunoprecipitation-based approaches has played a critical role in dissecting m6A functions and mechanisms of action. Yet, these approaches are of limited specificity, unknown sensitivity, and unable to quantify m6A stoichiometry. These limitations have severely hampered our ability to unravel the factors determining where m6A will be deposited, to which levels (the ‘m6A code’), and to quantitatively profile m6A dynamics across biological systems. Here, we used the RNase MazF, which cleaves specifically at unmethylated RNA sites, to develop MASTER-seq for systematic quantitative profiling of m6A sites at 16-25% of all m6A sites at single nucleotide resolution. We established MASTER-seq for orthogonal validation and de novo detection of m6A sites, and for tracking of m6A dynamics in yeast gametogenesis and in early mammalian differentiation. We discover that antibody-based approaches severely underestimate the number of m6A sites, and that both the presence of m6A and its stoichiometry are ‘hard-coded’ via a simple and predictable code within the extended sequence composition at the methylation sites. This code accounts for ~50% of the variability in methylation levels across sites, allows excellent de novo prediction of methylation sites, and predicts methylation acquisition and loss across evolution. We anticipate that MASTER-seq will pave the path towards a more quantitative investigation of m6A biogenesis and regulation in a wide variety of systems, including diverse cell types, stimuli, subcellular components, and disease states.
Leave a Comment